Jeśli zostanie zarejestrowany, aducanumab stanie się pierwszym lekiem redukującym postęp objawów klinicznych choroby Alzheimera, a także pierwszą terapią, która potwierdziłaby tezę, że usunięcie złogów amyloidu beta doprowadzi do uzyskania lepszych wyników klinicznych.
Zbliża się prawdziwy przełom w leczeniu choroby Alzheimera

Opublikowano 25 października 2019 13:04
Fot. Getty Images/iStockphoto
EMERGE (1638 pacjentów) oraz ENGAGE (1647 pacjentów) to wieloośrodkowe, randomizowane, porównawcze badania 3 fazy z podwójnie ślepą próbą i grupą kontrolowaną placebo, zaprojektowane w celu oceny skuteczności i bezpieczeństwa dwóch protokołów dawkowania aducanumabu.
Pierwszorzędowym celem badań była ocena skuteczności aducanumabu podawanego co miesiąc w porównaniu z placebo w ograniczaniu zaburzeń poznawczych oraz funkcjonalnych, których nasilenie mierzono za pomocą wyniku CDR-SB.
Drugorzędowym celem była ocena wpływu aducanumabu stosowanego co miesiąc w porównaniu z placebo na objawy kliniczne, które oceniano za pomocą MMSE, ADAS-Cog 13 oraz ADCS-ADL-MCI.
Badania te przerwano 21 marca 2019 roku po uzyskaniu wyników analizy nieskuteczności opartej na wcześniej określonych założeniach, która obejmowała węższy zestaw poprzednio zgromadzonych danych.
Analiza nieskuteczności opierała się na danych dostępnych w dniu 26 grudnia 2018 roku, zebranych w grupie 1748 pacjentów, którzy mieli możliwość uczestniczenia w całym 18-miesięcznym okresie badawczym i przewidywała, że po zakończeniu obu badań ich pierwszorzędowe punkty końcowe prawdopodobnie nie zostaną osiągnięte.
Warto przeczytać
Po przerwaniu EMERGE i ENGAGE udostępniono dodatkowe dane pochodzące z tych badań, które rozszerzyły zestaw danych, obejmując w sumie 3285 pacjentów, z których 2066 miało możliwość uczestniczenia w całej 18 miesięcznej terapii.
Nowa, obszerna analiza rozszerzonego zestawu danych wykazała wynik inny niż prognozowany przez analizę nieskuteczności. W szczególności, nowa analiza rozszerzonego zestawu danych wykazała istotność statystyczną wcześniej określonego pierwszorzędowego punktu końcowego badania EMERGE. Producent leku jest zdania, że podzbiór danych zgromadzonych w badaniu ENGAGE wspiera wyniki badania EMERGE, chociaż w badaniu ENGAGE nie osiągnięto pierwszorzędowego punktu końcowego. Firma skonsultowała się z zewnętrznymi doradcami oraz FDA w sprawie wspomnianych różnic w wynikach oraz ich konsekwencji.
– Ten obszerny [nowy] zestaw danych wykazuje po raz pierwszy fakt, że badanie 3 fazy potwierdziło, iż usunięcie złogów beta-amyloidu może ograniczyć zaburzenia o nasileniu klinicznym w przebiegu choroby Alzheimera i jest źródłem nowej nadziei dla przedstawicieli środowiska medycznego, pacjentów oraz ich rodzin – tłumaczy prof. Anton Porsteinsson, dyrektor Programu Opieki na Osobami z Chorobą Alzheimera, Badań oraz Edukacji (AD-CARE) Uniwersytetu w Rochester oraz główny badacz. – Istnieje ogromna, niezaspokojona potrzeba medyczna, społeczność związana z chorobą Alzheimera, czekała na ten moment. Namawiam firmę, FDA, społeczność medyczną oraz pacjentów, a także partnerów badań do wytrwałości w pracy, aby dzisiejsze oświadczenie stało się rzeczywistością – apeluje prof. Porsteinsson.
Pacjenci leczeni wysoką dawką aducanumabu w trakcie badania EMERGE, w którym osiągnięto wcześniej określony pierwszorzędowy punkt końcowy, wykazali znaczną redukcję postępu objawów klinicznych choroby względem punktu wyjściowego, które potwierdzono za pomocą CDR-SB w 78 tygodniu (23% w porównaniu z placebo). W badaniu EMERGE pacjenci leczeni wysoką dawką aducanumabu również wykazali podobne zmniejszenie nasilenia zaburzeń klinicznych, które mierzono za pomocą wcześniej określonych drugorzędowych punktów końcowych: MMSE (15% w porównaniu z placebo), ADAS-Cog 13 (27% w porównaniu z placebo) oraz ADCS-ADL-MCI (40% w porównaniu z placebo).
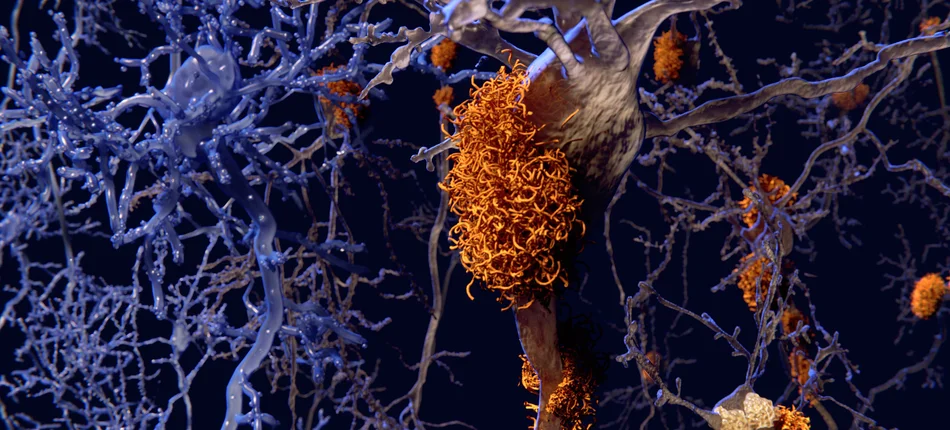
Obrazowanie nagromadzonych blaszek amyloidowych w badaniu EMERGE, wykazało, że nasilenie tego procesu uległo zmniejszeniu w porównaniu z placebo w 26 i 78 tygodniu, przy niskiej i wysokiej dawce aducanumabu. Te obserwacje kliniczne zostały potwierdzone przez dodatkowe dane dotyczące takiego biomarkera, jak stężenie białka tau w płynie mózgowo-rdzeniowym.
Po omówieniu danych podczas konsultacji z FDA firma jest przekonana, że różnice pomiędzy wynikami nowej analizy rozszerzonego zestawu danych a rezultatami analizy nieskuteczności były związane przede wszystkim z większą ekspozycją pacjentów na wysoką dawkę aducanumabu. W świetle nowej analizy rozszerzonego zestawu danych do większej ekspozycji na aducanumab przyczyniło się wiele czynników, w tym dane dotyczące większej liczby pacjentów, dłuższy średni czas trwania ekspozycji na wysoką dawkę, punkty w czasie, w których dokonywano zmian w protokole umożliwiających otrzymanie wysokiej dawki przez większy odsetek pacjentów, a także czas przeprowadzenia analizy nieskuteczności oraz jej wcześniej określone kryteria.
Uwzględniając wyniki dyskusji z FDA, firma planuje złożyć na początku 2020 roku wniosek o rejestrację w ścieżce Biologics License Application (BLA) oraz kontynuować rozmowę z urzędami regulacyjnymi na międzynarodowych rynkach, w tym w Europie i Japonii. Producent zamierza zaoferować możliwość kontynuowania leczenia aducanumabem pacjentom spełniającym warunki do włączenia, którzy wcześniej uczestniczyli w badaniach klinicznych 3 fazy, w długoterminowym badaniu stanowiącym przedłużenie badania PRIME fazy 1b oraz w badania EVOLVE, które dotyczyło oceny bezpieczeństwa.
Źródło: inf. pras.